Diagnosis: Carotid cavernous fistula
Discussion
Carotid Cavernous fistula (CCF) is a direct communication between the intracavernous portion of the carotid artery and the venous cavernous sinus. Usually is ipsilateral.
Most often results from significant trauma, penetrating or nonpenetrating.
Spontaneous rupture happens in the elderly.
Spontaneous rupture of CCF also associated with:
- Osteogenesis imperfecta
- Ehlers-Danlos syndrome
- Psuedoxanthoma elastica
Clinical presentation: acute onset pulsating exophthalmos, orbital bruit, dilated conjunctival vessels, and glaucoma. No pain whereas psuedotumor is almost always associated with pain.
Radiology
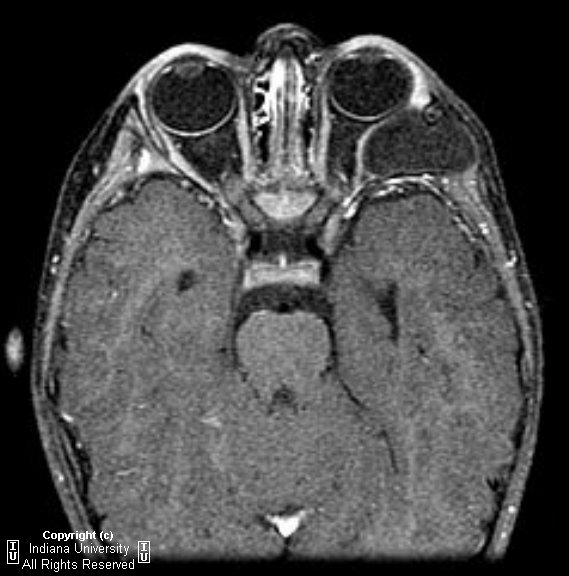
Unilateral diffuse enlargement of extra ocular muscles.
Proptosis.
Dilation of Superior ophthalmic vein (SOV) and venous structures within the carotid cavernous sinus due to backpressure. Our case was unusual in that the venous engorgement was on the opposite side of the CCF.
Irregularity of SOV may represent thrombus (not seen in this case).
Bowed convexity to the Cavernous sinus usually unilateral (our case demonstrated right cavernous sinus bowing due to the venous engorgement from the opposite carotid-very atypical)
US: reversal of flow in SOV.
Carotid Angio: filling of ophthalmic veins due to decompression of arterial flow with retrograde filling of orbital veins.
Repair: important to determine the extent of contralateral flow in the cavernous sinus without the CCF if carotid must be sacrificed. Glue embolization of the SOV and coiling are the primary treatments.
Diagnosis first by CT orbit findings then proceed to conventional angiogram for diagnosis and treatment.
Treatment: glue embolization and coiling. Approach varies but may go through the SOV from the internal jugular or direct approach by interventional neuroradiologist or neurosurgeon through the SOV as in our case.
Pearls
With carotid cavernous fistula look for enlarged superior opthalmic vein.(SOV) in addition to unilateral rectus enlargement and proptosis.
Need cerebral angiogram to definitively diagnose-may not see enlarged carotid cavernous sinus on CT.
SOV lives between the superior rectus and the optic nerve.
Treatment of CCF is with glue embolization and coiling