



Findings
There is expansion to the sella (Figure 1). No associated calcification is seen.
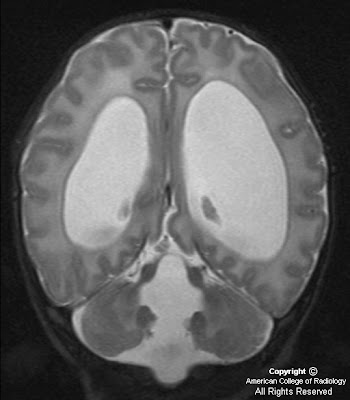
MR images demonstrate a lesion that is cystic and contains a fluid-fluid level with signal characteristics consistent with internal hemorrhage (Figure 4); the dependent fluid has signal characteristics consistent with acute blood. No nodularity or internal soft-tissue component is seen. The lesion exerts mass effect upon the optic chiasm, displacing it superiorly (Figure 3 and Figure 4). The infundibulum is displaced superiorly and anteriorly. The wall enhances (Figure 5 and Figure 7); there is no internal enhancement. There is no cavernous sinus invasion.
Diagnosis: Hemorrhagic Rathke's cleft cyst
In this case, craniopharyngioma was felt to be less likely since a high percentage of these calcify and typically display thick-walled, solid, or nodular enhancement. Macroadenoma was felt to be less likely due to lack of cavernous sinus invasion and the uniformly thin wall of this lesion.
Clinical
Rathke’s cleft cysts are often asymptomatic, but can enlarge and compress pituitary gland, hypothalamus, or optic tract.
Most common symptoms: hypopituitarism, visual disturbance, and headache.
Imaging
MRI appearance is variable; contents of cyst can be either simple or complex, secondary to blood or mucinous material. Thin wall may enhance, secondary to squamous metaplasia or peripherally displaced rim of pituitary tissue. Calcification is rare.