Findings
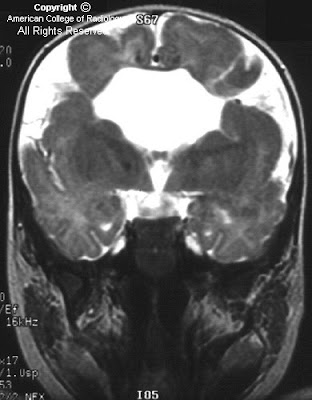
FindingsFigure 1: The image shows a fetus with only a single cerebral ventricle. Its thalami are fused. The findings are suggestive of holoprosencephaly, especially alobar.
Figure 2:The image of the anterior face shows a single nostril suggesting a midfacial anomaly. Holoprosencephaly is often associated with midline facial anomalies.
Diagnosis: HoloprosencephalyHoloprosencephaly is a malformation sequence involving the brain and often the face. It is a disorder of brain diverticulation resulting in partial or complete failure of cerebral hemisphere cleavage. Failure of cleavage results in failed formation of midline cranial structures. Midline brain development is associated with midface development and hence holoprosencephaly is associated with midface anomalies. Noncleavage of the primitive forebrain (the prosencephalon) leads to noncleavage in all planes. Associated olfactory and optic bulb anomalies occur because the prosencephalon, the primitive forebrain, does not cleave in a horizontal plane. A lack of proper cleavage in the transverse plane leads to improper formation of the telencephalon and diencephalon, which leads to abnormalities of thalamus and hypothalamus development. The often-seen fused or noncleaved thalami, as in the test case, may be a consequence. Finally, abnormal cleavage in a sagittal plane leads to abnormalities of the telencephalon, which normally forms the paired cerebral ventricles. In such patients an interhemisopheric fissure and other midline structures will not exist. The anomaly develops between the fourth and eighth week of embryonic life, well before the structures of the fetal brain can be adequately imaged by antenatal US. The abnormality is found in 1 in 16,000 live births, but in a larger number of fetuses and an even larger number of embryos (as high as 1 in 250), many of whom do not survive the pregnancy, hence affecting the numbers found among living births. There is a 12% recurrence rate for nonchromosomal cases.
Cases of holprosencephaly are divided into 3 forms. The most severe form, alobar holprosencephaly, typically results in a single holosphere (rather than 2 brain hemispheres), a single ventricle which often communicates with a dorsal sac, and fused thalami. There is no third or fourth ventricle, falx, corpus callosum, or interhemispheric fissure. The midbrain, brainstem, and cerebellum are typically normal unless made hypoplastic by mass effect of the large monoventricle or dorsal sac.
The intermediate form, ie, semilobar holoprosencephaly, shows at least partial prosencephalon cleavage. Temporal and occipital lobes may be separated. There may be a rudimentary interhemispheric fissure or a posterior falx. The incomplete form of holoprosencephaly, known as lobar holoprosencephaly, is the least devastating and may have a relatively normal appearing brain. There may simply be an absent septum pellucidum with an evident corpus callosum and perhaps fused or squared frontal horns. Rostral fusion may be the only ventricular abnormality with atria, occipital horns and temporal horns apparently normal.
When a child is born with a midline facial anomaly, a head US is performed to rule out holoprosencephaly. Facial abnormalities in cases of holoprosencephaly are variable. There may be none, or mild dysmorphism, such as hypotelorism or bilateral median cleft lip or palate. Cebocephaly, an intermediate form, has hypotelorism and a single nostrilled nose. Ethmocephaly is a more severe form of facial anomaly with severe hypotelorism, arhinia, and an interorbital single or double proboscis. Other severe abnormalities include cyclopia with variable eye and nose pairings. More significant facial anomalies are almost always associated with alobar holoprosencephaly.
Holprosencephaly is usually a terrible diagnosis no matter what its form. Those with severe brain abnormality, especially with arhinia or choanal atresia, die soon after birth if they survive the pregnancy. Less severely affected individuals may live several years but usually have severe neurologic and intellectual impairment. Even those with lobar holoprosencephaly who may live a normal lifetime may be severely retarded.