




Findings
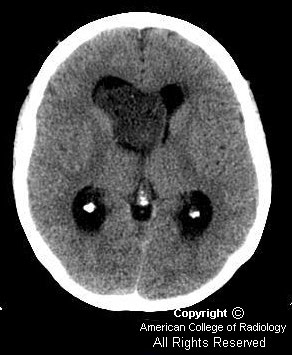
Noncontrast CT images show dilation of the lateral ventricles (Figure 1 and Figure 3) secondary to a large hypodense heterogeneous mass within the right lateral ventricle (Figure 2).
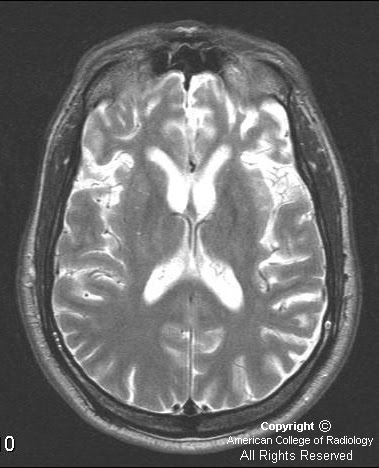
MRI brain with and without contrast. Images show a heterogeneous mass in the right lateral ventricle that causes obstruction of the foramen of Monroe and mild hydrocephalus. The mass is isointense to gray matter on T1 sequences (Figure 6), hyperintense on T2 (Figure 4), shows little if any enhancement on post contrast T1 (Figure 7), and is hyperintense with multiple cystic spaces on FLAIR images (Figure 5).
Differential diagnoses: Intraventricular tumors
- Meningioma
- Choroid plexus papilloma or carcinoma
- Ependymoma
- Subependymoma
- Central neurocytoma
- Metastasis
- Subependymal giant cell astrocytoma
Diagnosis: Subependymoma
Subependymomas are benign intraventricular tumors that arise from cells of the subependymal plate, including subependymal glia, astrocytes and ependymal cells. They are rare, accounting for less than 1% of all intracranial neoplasms. They can occur with ependymomas, which can worsen prognosis. They occur in either the lateral or fourth ventricles, being attached to either septum pellucidum or wall of lateral ventricle, or the ventricular floor, respectively. The larger the lesion, the more likely cystic change and calcification are encountered; however, the calcification is more often punctate and fine rather than coarse, as seen in other intraventricular tumors. Mean age of presentation is 60 years in asymptomatic patients, but only 40 years if symptomatic. There is a male predominance. Surgical resection is often curative, although fourth ventricular subependymomas pose a challenge at total resection.
On CT, an iso- to hypodense intraventricular mass will be seen with cystic change and calcification in larger lesions, and associated hydrocephalus if obstructing. Mild to no enhancement after contrast is typical. On MR, subependymomas are T2/FLAIR hyperintense and T1 isointense with mild to no enhancement with contrast. Because fourth ventricular subependymomas can have calcification and contrast enhancement more typical of ependymomas, diagnostic imaging is more challenging.
Another diagnostic challenge on imaging is the central neurocytoma, a recently described tumor that was routinely mistaken for an intraventricular oligodendroglioma on histologic analysis. These intraventricular tumors have a striking resemblance to a subependymoma on CT and MR images. Neurocytomas, however, more reliably enhance after contrast administration, and have a more heterogeneous cystic appearance. They also are usually found in younger patients.